In a class of roughly 30 neurological disorders that includes ALS, Huntington’s Disease and Fragile X Syndrome, the relevant mutant gene features sections of repeating base pair sequences known as short tandem repeats, or STRs. Healthy people have STRs of normal lengths distributed across their DNA. For people with trinucleotide repeat (TNR) expansion diseases, however, the STRs in the mutant genes are unstable: the number of repetitions expand into ultra-long lengths that are related to the disease’s pathology.
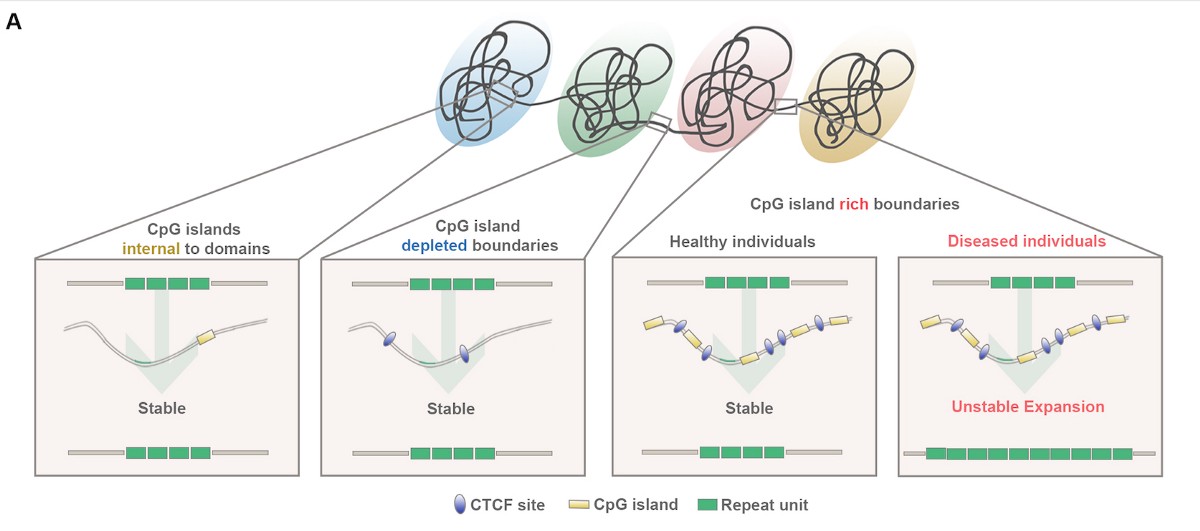
Researchers at the University of Pennsylvania have found another common thread linking nearly all of the TNR expansion diseases: the complicated 3D patterns that the DNA is folded into in order to fit in the nucleus of the cell. They found that nearly all of the STRs known to grow unstable in disease are located at the boundaries that separate neighboring folded domains.
The researchers also created high-resolution genome folding maps around the FMR1 gene in patients with Fragile X Syndrome and in healthy individuals. They found that the 3D genome was misfolded in disease; the boundary was destroyed around FMR1 in all Fragile X patients who also had pathologic STR expansion and FMR1 silencing.
The findings establish a strong correlation between 3D genome misfolding, short tandem repeat instability, and pathologic gene disruption in these deadly and debilitating diseases, suggesting new research questions whose answers could improve diagnosis or treatment.

The research was led by Jennifer E. Phillips-Cremins, assistant professor in Penn Engineering’s Department of Bioengineering and in the Perelman School of Medicine’s Department of Genetics, along with James Sun and Linda Zhou, members of her lab.
Their study was published in the journal Cell.
At the core of the mystery behind TNR diseases is what causes their repeating sequences to pathologically expand. The same exact repeating patterns appear in hundreds of thousands of other locations along the linear genome, in genes and non-coding regions alike, but are not known to become unstable.
“I wanted to work on this project because I was fascinated by the idea that 3D genome folding — something I had barely even heard of before joining the Cremins lab — could be the missing piece to understanding why certain parts of the genome behave the way they do,” Zhou says. “In this case, why can repetitive DNA in certain genes become unstable while others do not?”
Mapping the DNA’s 3D folding patterns has only recently become possible, so the genes related to TNR diseases have predominantly been studied via their linear sequences.
The researchers’ findings were enabled by the Cremins Lab’s technique for producing genome folding maps. By fixing the DNA such that its 3D folding patterns are preserved prior to sequencing, two distant parts of the linear sequence will end up in the same string of hybrid DNA and will thus be detected together when the DNA is sequenced. Statistically mapping these associations provides a high-definition picture of which disparate parts of a linear sequence are in physical contact — and thus can affect one another’s gene expression — when the genome is in its folded state.
Zoomed out, this map looks like a line of densely packed cities, set off from one another by stretches of highway. Each of these “cities” is a topologically associated domain, or TAD, and contains sequences that are in physical contact with one another. The highways between the cities are known as boundaries; they contain linker sequences that physically prevent neighboring TADs from interacting with one another.
In trying to determine what makes the unstable repeats associated with TNR diseases different from their stable counterparts, the researchers considered whether their location with respect to genome folding patterns played a role.
“Every human individual has hundreds of thousands of short tandem repeat tracts distributed throughout their genome. The repeats exhibit wide variation in sequence, location in the gene body, normal and mutation length ranges, the cell types they affect and the phenotypes they produce,” Phillips-Cremins says. “But, for the handful of short tandem repeat tracts known to grow unstable in disease, nearly all are localized specifically to genome folding boundaries.”
The strength of this correlation immediately raised questions of causality for the researchers: do boundaries provide a local environment where the genome is susceptible to repeats, or do the repeats determine the location of a boundary in some way?
“Recent studies have provided strong evidence that the 3D genome can be misfolded in specific cancers and disorders of limb development,” Phillips-Cremins says. “As we initiated our studies to understand the cause-and-effect relationship between boundary placement and repeat expansion, we also began to wonder if we would observe topological changes in TNR diseases or if the boundaries remained intact at the locations of repeat instability.”
Collaborating with Beverly Davidson of the Children’s Hospital of Philadelphia and Flora Tassone of UC Davis, Phillips-Cremins and her co-authors studied brain tissue and B cells donated from patients with Fragile X syndrome and compared them to cells donated by their healthy siblings.
“We discovered that in Fragile X Syndrome, the leading cause of intellectual disability in the United States, the DNA is misfolded at precisely the same location where the genetic defect and pathogenic gene silencing occurs,” Sun says. “This finding raises the profile of many important questions about the relationship between genome folding, repeat expansion, and gene silencing, which we hope will one day lead to insights that might inspire therapeutic options for children affected with the disease.”
That the boundary home to the Fragile X gene is destroyed is highly suggestive that this change is related to the disease’s gene silencing. Questions of causality will need to be addressed before researchers fully understand folding’s role. The recently developed gene editing technology CRISPR/Cas9 will enable Phillips-Cremins and her colleagues to conduct experiments that explore the potential causal role of the 3D genome in repeat expansion.
“We’re tremendously excited to examine whether and how the 3D genome misfolding in Fragile X syndrome is causally linked to the silencing of gene expression in this disease. In our ongoing studies, we want to determine if gene silencing disrupts the boundary, or if boundary disruption silences the gene,” Phillips-Cremins says. “Mechanistic studies have predominantly focused on the linear DNA thus far, but we can now add a new third dimension to understanding the genetic underpinnings of TNR diseases.”
Cremins Lab members Daniel Emerson, Sai Phyo, Katelyn Titus, Wanfeng Gong, Thomas Gilgengast and Jonathan Beagan also contributed to this study.
This research was funded by The New York Stem Cell Foundation, the Alfred P. Sloan Foundation, the National Institutes of Health through a NIH Director’s New Innovator Award (1DP2MH11024701), a 4D Nucleome Common Fund grant (1U01HL12999801), a research project grant (R01 GM113929), and a joint National Science Foundation — National Institute of General Medical Sciences grant to support research at the interface of the biological and mathematical sciences (1562665). Phillips-Cremins is a New York Stem Cell Foundation (NYSCF) Robertson Investigator and an Alfred P. Sloan Foundation Fellow.